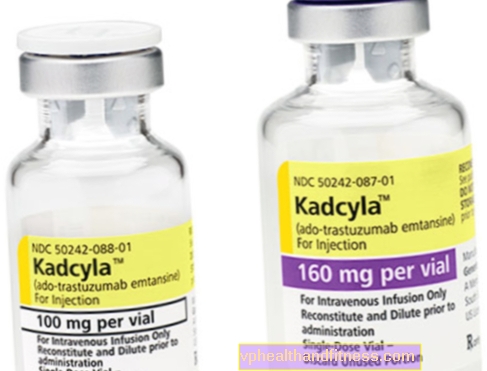
1 флакон съдържа 100 mg (160 mg) прах за разтваряне 5 ml (8 ml) трастузумаб емтанзин концентрат в концентрация 20 mg / ml за инфузия след разтваряне.
| Име | Съдържание на пакета | Активното вещество | Цена 100% | Последно модифициран |
| Кадцила | 1 флакон, прах за приготвяне окончателно решение до инф. | Трастузумаб емтанзин | 2019-04-05 |
Действие
Противораково лекарство. Трастузумаб емтанзин е конюгат антитяло-лекарство, който съдържа трастузумаб, хуманизирано анти-HER2 IgG1 моноклонално антитяло, ковалентно свързано с DM1 (производно на матанзин, инхибитор на микротубули) чрез стабилна MCC тиоетерна връзка (4- циклохексан-1-карбоксилат) . Emtansine е MCC-DM1 комплекс. Конюгацията на DM1 с трастузумаб причинява селективно действие на цитотоксичните лекарства срещу свръхекспресиращи туморни клетки HER2, като по този начин увеличава вътреклетъчната концентрация на DM1 директно в туморните клетки. При свързване с HER2, трастузумаб емтанзин е медиирана от рецептора интернализация, последвана от лизозомно разграждане, освобождавайки DM1-съдържащи катаболити (главно лизин-MCC-DM1). Механизмът на действие на трастузумаб емтанзин се медиира от активността на трастузумаб и DM1. Трастузумаб емтанзин, подобно на трастузумаб, се свързва с домейн IV на извънклетъчния домен (ECD) на рецептора, както и с Fcγ рецептори и допълва C1q. Освен това, той инхибира активността на ECD домена на HER2 рецептора, инхибира сигнализирането на фосфатидилинозитол 3-киназата (PI3-K) и медиира антитяло зависима клетъчна цитотоксичност (ADCC) в човешките клетки на рак на гърдата, свръхекспресиращи HER2. DM1 се свързва с тубулин. Чрез инхибиране на полимеризацията на тубулина, както DM1, така и трастузумаб емтанзин предизвикват спиране на клетките във фаза G2 / M на клетъчния цикъл, което в крайна сметка води до клетъчна смърт чрез апоптоза. MCC линкерът намалява системното освобождаване на DM1 и увеличава концентрацията му в целевото място. Трастузумаб емтанзин се деконюгира и след това се катаболизира чрез протеолиза в клетъчните лизозоми. DM1 се метаболизира предимно от CYP3A4 и в по-малка степен от CYP3A5. T0.5 на трастузумаб е приблизително 4 дни. Не е имало натрупване на трастузумаб емтанзин след множество интравенозни инфузии, прилагани на интервал от 3 седмици. Възрастта няма ефект върху фармакокинетиката на трастузумаб емтанзин.
$config[ads_text1] not found
Дозировка
Интравенозно. Препаратът трябва да се предписва от лекар и да се прилага под наблюдението на здравни специалисти с опит в лечението на пациенти с рак. Пациентите, получаващи трастузумаб емтанзин, трябва да имат HER2-позитивен рак - имунохистохимичен (IHC) резултат 3+ или съотношение ≥2 при in situ хибридизация (ISH). Тестовете трябва да се извършват с помощта на in vitro диагностични тестове (IVD) с CE маркировка. Ако CE IVD тестът не е наличен, тестът трябва да се извърши с друг валидиран тест. За да се избегнат медицински грешки, е важно да проверите етикета на флакона, за да се уверите, че лекарството, което се приготвя и прилага, е Kadcyla (трастузумаб емтанзин), а не Herceptin (трастузумаб). Препоръчителната доза е 3,6 mg / kg телесно тегло. дава се под формата на интравенозна инфузия на всеки 3 седмици (21-дневен цикъл). Пациентите трябва да бъдат лекувани до прогресиране на тумора или постигане на неприемлива токсичност. Началната доза трябва да се прилага като 90-минутна интравенозна инфузия. Мястото на инжектиране трябва да бъде внимателно наблюдавано поради възможността за подкожно проникване на лекарството по време на приложението. Ако предишната инфузия се понася добре, следващите дози могат да се дадат като 30-минутна инфузия. Пациентите трябва да бъдат наблюдавани по време на инфузията и поне 30 минути след това. Скоростта на инфузия трябва да бъде намалена или прекъсната, ако пациентът изпитва симптоми, свързани с инфузията. Трастузумаб емтанзин трябва да се прекрати в случай на животозастрашаващи инфузионни реакции. Лекарствата за алергични / анафилактични инфузионни реакции, както и спешно оборудване трябва да бъдат на разположение за незабавна употреба. Ако се пропусне планирана доза, тя трябва да се приложи възможно най-скоро; не чакайте следващия цикъл. Схемата на дозиране трябва да бъде коригирана, за да се поддържа 3-седмичният интервал на дозиране. Следващата доза трябва да се прилага съгласно препоръките за дозиране. Управлението на симптоматичните нежелани реакции може да включва периодично спиране, намаляване на дозата или прекратяване на лечението. График за намаляване на дозата (начална доза 3,6 mg / kg): първо намаляване на дозата 3 mg / kg; второ намаляване на дозата 2,4 mg / kg т.т .; ако е необходимо допълнително намаляване на дозата, лечението трябва да бъде спряно. Насоки за промяна на дозата за повишаване на AST и ALT: Степен 2 (> 2,5 до ≤5 x ULN) - не е необходимо коригиране на дозата; степен 3 (> 5 до ≤20 x ULN) - използвайте препарата, ако AST / ALT намалява до степен ≤ 2 (> 2,5 до 20 x ULN) - прекратете лечението. Правила за промяна на дозата при хипербилирубинемия: степен 2 (> 1,5 до ≤3 x ULN) - използвайте препарата, когато нивото на билирубина е спаднало до степен ≤1. (> ULN до 1,5 x ULN), не е необходимо коригиране на дозата; Степен 3 (> 3 до ≤10 x ULN) - използвайте препарата, ако концентрацията на билирубин намалее до степен ≤ 1 (> ULN до 1,5 x ULN), след това намалете дозата (вижте графика за намаляване на дозата); Степен 4 (> 10 x ULN) - Крайно лечение. Правила за промяна на дозата при тромбоцитопения: степен 3 (концентрация на тромбоцитите 25 000 до 3) - използвайте препарата, когато концентрацията на тромбоцитите е ≤1. (напр. ≥75 000 / mm3); не е необходимо коригиране на дозата; Степен 4 (концентрация на тромбоцитите 3) - използвайте препарата, когато концентрацията на тромбоцитите достигне ≤1. (напр. ≥75 000 / mm3) след това намалете дозата (вижте графика за намаляване на дозата). Принципи на промяна на дозата при пациенти с камерна дисфункция: LVEF 45% - продължете терапията; LVEF 40% до ≤45% с едновременно намаляване на фракцията на изтласкване с - не използвайте препарата; Преоценка на LVEF в рамките на 3 седмици, прекратете терапията, ако LVEF все още е ≥10 процентни пункта от изходното ниво; симптоматична СНС - спиране на лечението. Деца и юноши: Безопасността и ефикасността при деца и юноши под 18-годишна възраст не са установени, тъй като дисеминираният рак на гърдата (MBC) не е известен при тази популация. Специални групи пациенти. Терапията трябва да бъде временно прекъсната при пациенти, които развият периферна невропатия 3 или 4 степен, докато достигне степен ≤2; при възобновяване на лечението може да се обмисли намаляване на дозата, както е посочено в схемата за намаляване на дозата. Не е необходимо коригиране на дозата при пациенти на възраст ≥ 65 години; няма достатъчно данни за определяне на безопасността и ефикасността на терапията при пациенти на възраст ≥75 години. Не се налага корекция на началната доза при пациенти с леко или умерено бъбречно увреждане; Потенциалната нужда от корекция на дозата при пациенти с тежка бъбречна недостатъчност не може да бъде определена и поради това тези пациенти трябва да бъдат внимателно наблюдавани. Не е необходимо коригиране на началната доза при пациенти с леко или умерено чернодробно увреждане. Трастузумаб емтанзин не е проучван при пациенти с тежко чернодробно увреждане; трябва да се внимава при лечение на пациенти с чернодробно увреждане поради известна хепатотоксичност, наблюдавана по време на лечението. Начин на даване. Препаратът трябва да се разтваря и разрежда от медицинския персонал и да се прилага като интравенозна инфузия. Не трябва да се прилага като болус или бърза инжекция.
$config[ads_text2] not foundПоказания
Монотерапия на възрастни пациенти с HER2-позитивен, неоперабилен локално напреднал или метастатичен рак на гърдата, лекуван преди това с трастузумаб и таксан, в комбинация или поотделно. Пациенти, които са имали предишно лечение за локално напреднало или генерализирано заболяване или са имали рецидив по време на или в рамките на 6 месеца след завършване на адювантната терапия.
Противопоказания
Свръхчувствителност към активното вещество или други съставки на препарата.
Предпазни мерки
В клинични проучвания с трастузумаб емтанзин са докладвани случаи на интерстициална белодробна болест (ILD), включително пневмония; някои са свързани с остра дихателна недостатъчност или са били фатални. Прекратяване на лечението с препарата се препоръчва при пациенти с диагноза ILD или пневмония. Пациентите с диспнея в покой, свързани с усложнения на напреднал рак и свързани заболявания могат да бъдат изложени на повишен риск от развитие на белодробни усложнения. По време на лечението с препарата се наблюдават хепатотоксичност и тежки нарушения на черния дроб и жлъчните пътища, включително нодуларна регенеративна хипертрофия (NRH) на черния дроб и смъртни случаи, причинени от медикаментозно увреждане на черния дроб; съпътстващи заболявания и / или съпътстващи лекарства, за които е известно, че имат хепатотоксичен потенциал, също могат да бъдат засегнати. Чернодробната функция трябва да се проследява преди започване на лечението и преди последващото дозиране. Пациентите с изходно повишение на ALT (напр. Свързани с метастази в черния дроб) са предразположени към развитие на чернодробна недостатъчност и са изложени на по-висок риск от чернодробна токсичност степен 3-5 или повишени чернодробни лабораторни нива. Случаите на NRH се диагностицират чрез вземане на чернодробни биопсии. Появата на NRH трябва да се има предвид при всички пациенти с клинични признаци на портална хипертония и / или подобно на цироза изображение, наблюдавано при компютърна томография на черния дроб, но с нормални нива на серумна трансаминаза и без други доказателства за цироза. Ако се диагностицира NRH, лечението с препарата трябва да се прекрати. Не е проучен при пациенти със серумни трансаминази> 2,5 x ULN (горна граница на нормата) или с общ билирубин> 1,5 x ULN преди започване на лечението. В случай на активност на серумни трансаминази> 3 x ULN и едновременно обща концентрация на билирубин> 2 x ULN, лечението трябва да бъде прекратено. Трябва да се внимава при лечение на пациенти с чернодробно увреждане. Има повишен риск от левокамерна дисфункция по време на лечението. При пациенти, лекувани с трастузумаб емтанзин, се наблюдава намаляване на фракцията на изтласкване на лявата камера (LVEF) на 50 години), с първоначална ниска стойност на LVEF (25 kg / m2). Преди започване на лечението и на редовни интервали (напр. На всеки 3 месеца) трябва да се извършват стандартни тестове за сърдечна функция (ехокардиограма или многозатворна радиоизотопна ангиография). LVEF на пациентите на изходно ниво е ≥50% в повечето клинични проучвания. Пациенти със застойна сърдечна недостатъчност, тежки аритмии, изискващи лечение, анамнеза за миокарден инфаркт или нестабилна стенокардия през 6-те месеца преди рандомизирането или с диспнея в покой поради напреднала неопластична болест са изключени от проучването. В случай на левокамерни нарушения, прилагането на следващата доза трябва да бъде отложено или терапията да бъде прекратена. Ефектът от лечението с трастузумаб емтанзин при пациенти, прекъсващи лечението с трастузумаб поради реакция, свързана с инфузия, не е проучен; терапията с това лекарство не се препоръчва при тази група пациенти. Терапията трябва да бъде прекратена при пациенти, които развият тежка реакция, свързана с инфузията, докато симптомите отзвучат. Повторното започване на терапията трябва да се обмисли въз основа на клиничната преценка за тежестта на реакцията. Лечението трябва да се прекрати в случай на животозастрашаваща инфузионна реакция. Ефектът от лечението с трастузумаб емтанзин при пациенти, преустановени от приема на трастузумаб поради свръхчувствителност, не е проучен; Употребата на трастузумаб емтанзин при тези пациенти не се препоръчва. Пациентите трябва да бъдат наблюдавани за свръхчувствителност / алергични реакции, симптомите могат да бъдат същите като реакциите, свързани с инфузията; са наблюдавани тежки анафилактични реакции. Ако се появи свръхчувствителност (с повишени реакции по време на последващи инфузии), лечението с трастузумаб емтанзин трябва да бъде прекратено.Поради риска от хеморагични събития (включително ЦНС, респираторен и стомашно-чревен кръвоизлив), трябва да се внимава и да се обмисли допълнително наблюдение при едновременно приложение с антикоагуланти или антитромбоцитни лекарства. Препоръчва се броят на тромбоцитите да се проследява преди прилагането на всяка доза трастузумаб емтанзин. Пациенти с тромбоцитопения (≤100 000 / mm3) и пациенти, получаващи антикоагулантна терапия (напр. Варфарин, хепарин, нискомолекулни хепарини), трябва да бъдат внимателно наблюдавани по време на лечението с трастузумаб емтанзин. Трастузумаб емтанзин не е проучван при пациенти с брой тромбоцити ≤ 100 000 / mm3 преди започване на терапията. В случай на влошаване на тромбоцитопенията до степен 3 или по-висока (3), препаратът не трябва да се използва, докато токсичността не намалее до степен 1 (≥75 000 / mm3). В клинични проучвания с трастузумаб емтанзин се наблюдава периферна невропатия степен 1, главно сензорна. Пациенти със степен ≥3 периферна невропатия бяха изключени от участие в клинични изпитвания. Лечението на пациенти с периферна невропатия от степен 3 или 4 трябва периодично да се прекъсва, докато усложнението се намали до степен ≤2. Пациентите трябва да се наблюдават редовно за признаци на невротоксичност. За да се подобри проследимостта на биологичните препарати, търговското наименование на прилаганото лекарство трябва да бъде ясно написано (или посочено) в досието на пациента. Безопасността и ефикасността на лекарството при деца и юноши под 18-годишна възраст не са установени, тъй като при тази популация не се открива разпространен рак на гърдата. Не трябва да се прилага като болус или бърза инжекция.
$config[ads_text3] not found
Нежелана дейност
Много чести: инфекция на пикочните пътища, тромбоцитопения, анемия, хипокалиемия, безсъние, периферна невропатия, главоболие, кървене, епистаксис, кашлица, диспнея, стоматит, диария, повръщане, гадене, запек, сухота в устата, коремна болка , обрив, мускулно-скелетна болка, артралгия, миалгия, умора, треска, астения, втрисане, повишаване на трансаминазите. Чести: неутропения, левкопения, свръхчувствителност, замаяност, дисгевзия, увреждане на паметта, синдром на сухото око, конюнктивит, зрителни нарушения, повишена лакримация, левокамерна дисфункция, хипертония, диспепсия, гингивално кървене, кожен сърбеж, алопеция, заболяване на ноктите , палмарно-плантарна еритродизестезия, уртикария, периферен оток, повишени нива на алкална фосфатаза, реакции, свързани с инфузията (зачервяване на кожата, студени тръпки, треска, диспнея, ниско кръвно налягане, хрипове, бронхоспазъм, тахикардия). Нечести: пневмония (ILD), хепатотоксичност, чернодробна недостатъчност, нодуларна регенеративна хипертрофия, портална хипертония, екстравазация на мястото на инжектиране (еритем, чувствителност, дразнене на кожата, болка или подуване). Освен това се наблюдава хипербилирубинемия. В клинични проучвания повишенията в серумните трансаминази (степен 1-4) са настъпили по време на лечението с трастузумаб емтанзин и обикновено са били преходни. Повишаването на трансаминазите е най-често преходно, достигайки връх на 8-ия ден след дозиране. След това токсичността намалява до степен 1 или отзвучава преди следващия цикъл. Налице е и кумулативен ефект (процентът на пациентите с повишаване на степен ALT / AST степен 1-2 се увеличава с следващите цикли). При пациенти с повишени трансаминази, по-голямата част от пациентите са имали намаляване на токсичността или възстановяване на степен 1 в рамките на 30 дни след последната доза трастузумаб емтанзин. Дисфункция на лявата камера е установена при 2,2% от пациентите, участващи в клинични изпитвания. В повечето случаи намаленията на LVEF от степен 1 или 2 са асимптоматични.Токсичност от степен 3 или 4 се наблюдава при 0,4% от пациентите, обикновено по време на началните цикли на лечение (1-2). Тежки (степен ≥3) хеморагични събития са съобщени при 2,2% от всички пациенти. Тромбоцитопения се наблюдава при 24,9% от пациентите в клинични изпитвания и е най-честата нежелана реакция, водеща до прекратяване на терапията. В клинични проучвания честотата и тежестта на тромбоцитопенията са по-високи при азиатски пациенти. Някои пациенти, които са развили тези усложнения, също са лекувани с антикоагулант. Имаше смъртоносни епизоди на кървене и тежки усложнения от кървене, включително кървене от ЦНС. 5.3% от пациентите са тествани положително за антитела срещу трастузумаб емтанзин.
$config[ads_text4] not foundБременност и кърмене
Не се препоръчва употребата на трастузумаб емтанзин при бременни жени. Преди да забременеят, жените трябва да бъдат информирани за възможността да навредят на плода. Въпреки това пациентите, които забременеят, трябва незабавно да се свържат с лекар. Ако бременна жена се лекува с трастузумаб емтанзин, се препоръчва внимателно наблюдение от мултидисциплинарен екип. Трастузумаб може да причини увреждане или смърт на плода, ако се прилага на бременна жена. При деца на бременни жени, лекувани с препарата, са докладвани случаи на олигохидрамнион, някои от които водят до фатална белодробна хипоплазия. DM1, цитотоксичният компонент на трастузумаб емтанзин, може да бъде тератогенен и потенциално ембриотоксичен. Жените трябва да спрат кърменето преди започване на терапия с трастузумаб емтанзин. Пациентите могат да започнат да кърмят 7 месеца след спиране на лечението. Жените с детероден потенциал трябва да използват ефективна контрацепция по време на лечението с лекарството и в продължение на 7 месеца след последната доза трастузумаб емтанзин. Ефективните методи за контрацепция също трябва да се използват от мъже или техните партньори.
Коментари
Пациентите, които изпитват реакции, свързани с инфузията, не трябва да шофират или да работят с машини, докато тези симптоми отзвучат. Информация, изготвена на базата на SPC от 13 юли 2017 г. Текущият КХП е достъпен на www.roche.pl.
Взаимодействия
Резултатите от in vitro изследвания на метаболизма в човешките чернодробни микрозоми показват, че DM1 се метаболизира предимно от ензима CYP3A4 и в по-малка степен от CYP3A5. Трябва да се избягва едновременната употреба на силни инхибитори на CYP3A4 (например кетоконазол, итраконазол, кларитромицин, атазанавир, индинавир, нефазодон, нелфинавир, ритонавир, саквинавир, телитромицин и вориконазол), тъй като това може да повиши нивата на DM1 и токсичността. Трябва да се обмисли алтернативна формулировка, която не инхибира или само леко CYP3A4. Ако едновременната употреба на силни инхибитори на CYP3A4 не може да бъде избегната и, когато е възможно, помислете за отлагане на приложението на трастузумаб емтанзин, докато инхибиторите на CYP3A4 се изчистят от циркулацията (приблизително 3 полуживота на инхибиторите). Ако обаче се използва едновременно силен инхибитор на CYP3A4 и лечението с трастузумаб емтанзин не може да бъде отложено, пациентът трябва да бъде внимателно наблюдаван за нежелани ефекти в такива случаи.
Препаратът съдържа веществото: Трастузумаб емтанзин
Възстановено лекарство: НЕ





-porada-eksperta.jpg)